Overview
The rational discovery, study, design, and optimisation of chemicals and materials are central to scientific, technological, economic, and societal progress, but are limited by long, expensive research and development cycles relying on trial and error. Advances in artificial intelligence and machine learning algorithms and their applications promise significant acceleration by enabling predictions and simulations of complex material properties and processes in quantitative agreement with experiments.
We develop data-driven methods to solve problems in physics, chemistry, materials science, and adjoining fields. Bridging the divide between experiments and simulations, we pioneer the accelerated discovery, study, design, and optimisation of novel materials, chemicals, and their processes through data-driven predictions, advanced simulations, and automated experimental platforms. Our contributions range from fundamental science to solutions for real-world challenges.
Our focus is on accurate, computationally efficient machine-learning surrogate models of expensive-to-evaluate functions, such as results of wet-lab experiments or electronic-structure calculations. Our research includes machine-learning interatomic potentials for accurate all-atom simulations at unprecedented time and length scales and multi-objective surrogate-based (Bayesian) optimisation for property prediction and the design of chemicals and materials.
Expertise
Machine learning methods we use include
- Linear methods (e.g., least-squares regression, principal component analysis)
- Kernel methods (e.g., kernel ridge regression, Gaussian processes, support vector machines, kernel principal component analysis)
- Artificial neural networks (e.g., feedforward networks, convolutional networks, diffusion models)
- Deep learning (e.g., message-passing networks, equivariant networks)
- Learning with derivatives (e.g., machine-learning interatomic potentials)
- Other methods (e.g., subgroup discovery, symbolic regression)
Atomistic systems we investigate include
- Elements (e.g., tungsten, warm dense hydrogen)
- Crystals and solids (e.g., zirconia, borosilicate glasses)
- Surfaces and interfaces (e.g., titania, alumina)
- Polymers and composite materials (e.g., vitrimers, flax fibre biocomposites)
- Small organic molecules (in vacuum and in solvents)
- Drugs, drug-like molecules, and biomolecules (e.g., truxillic acid derivatives, triazoles, archazolid A)
Properties and processes we study include
- Phase transitions (e.g., in H under pressure)
- Heat transport (e.g., via the Green-Kubo formalism)
- Nuclear chemical shifts (e.g., in organic molecules)
- Catalytic reactions (e.g., CO2 reduction)
- Deposition processes (e.g., gold on alumina)
- Drug-target binding (e.g., PPARγ, Farnesoid X, cyclooxygenase-1 receptors)
Features we use include
- Local atomic environment descriptions (e.g., Coulomb matrix, many-body tensor, many-body expansions, atomic cluster expansion)
- Molecular structure graphs (e.g., descriptors, fingerprints, pharmacophores, graph kernels)
- Experimental measurements (e.g., of acoustic emissions, from ellipsometry)
Examples
Machine-learning interatomic potentials (MLIPs) accelerate molecular dynamics simulations by several orders of magnitude compared to the underlying ab initio reference calculations. This enables accurate simulations at unprecedented time and length scales. We develop ultra-fast potentials [xrh23], the currently fastest MLIPs; contribute to the validation and benchmarking of MLIPs [bjr24, pcmt25, ppmt2025]; and apply MLIPs to study atomistic systems and their processes [tjrc25].
In quantum mechanics, we demonstrated for the first time that accurate prediction of atomisation energies across chemical compound space is possible. [rtml12] This was enabled by representations [lgr22], first the Coulomb matrix, and later the many-body tensor representation [hr22]. Follow-up publications [hmtm13] include the Δ-learning approach [rdrl15] and the widely used QM9 dataset [rdrl14].
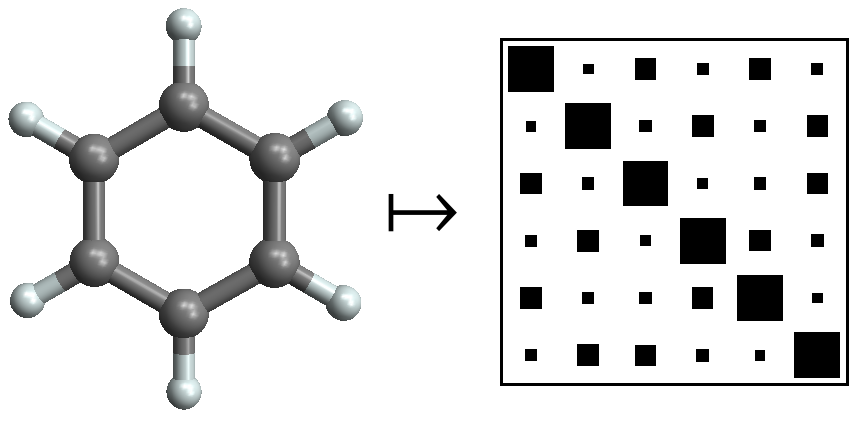
In density functional theory, we laid the foundation for machine-learning functionals by demonstrating that machine learning can approximate density functionals. [srmb12] We further developed this line of research in follow-up work. [srmb13, srmb15, vsmb15, lsmb15] It has since become a research direction of its own.
In physical chemistry, we optimised transition state theory dividing surfaces with machine learning [phmh12]. We predicted acid dissociation constants (pKa values) of monoprotic compounds [rkt11, rkt10] with kernel regression and graph kernels [rs10] tailored for small organic molecules [rps07], as well as nuclear chemical shifts [rrl15] in organic molecules.
In medicinal chemistry, we identified a novel agonist of the diabetes-related transcription factor PPARγ (peroxisome proliferator-activated receptor γ) with Gaussian process-based virtual screening and cellular reporter gene assays. [rsms10] We investigated the identified truxillic acid derivative in follow-up studies. [srss10, ssss10]